
Entropy is rather intimidating. It’s important to the sciences of physics and chemistry but it’s also highly abstract. There are, no doubt, more than a couple of students who graduate with college degrees in the physical sciences or in engineering who don’t have much of an understanding of what it is or what to do with it. We know it’s there and that it’s a thing but we’re glad not to have to think about it any more after we’ve crammed for that final exam in thermodynamics. I think one reason for that is because entropy isn’t something that we often use. And using things is how we come to understand them, or at least get used to them.
Ludwig Wittgenstein argued in his later philosophy that the way we learn words is not with definitions or representations but by using them, over and over again. We start to learn “language games” as we play them, whether as babies or as graduate students. I was telling my daughters the other day that we never really learn all the words in a language. There are lots of words we’ll never learn and that, if we happen to hear them, mean nothing to us. To use a metaphor from Wittgenstein again, when we hear these words they’re like wheels that turn without anything else turning with them. I think entropy is sometimes like this. We know it’s a thing but nothing else turns with it. I want to plug it into the mechanism. I think we can understand entropy better by using it to solve physical problems, to see how it interacts (and “turns”) with things like heat, temperature, pressure, and chemical reactions. My theory is that using entropy in this way will help us get used to it and be more comfortable with it. So that maybe it’s a little less intimidating. That’s the object of this episode.
I’ll proceed in three parts.
1. Define what entropy is
2. Apply it to problems using steam
3. Apply it to problems with chemical reactions
What is Entropy?
I’ll start with a technical definition that might be a little jarring but I promise I’ll explain it.
Entropy is a measure of the number of accessible microstates in a system that are macroscopically indistinguishable. The equation for it is:
S = k ln W
Here S is entropy, k is the Boltzmann constant, and W is the number of accessible microstates in a system that are macroscopically indistinguishable.
Most people, if they’ve heard of entropy at all, haven’t heard it described in this way, which is understandable because it’s not especially intuitive. Entropy is often described informally as “disorder”. Like how your bedroom will get progressively messier if you don’t actively keep it clean. That’s probably fine as an analogy but it is only an analogy. I prefer to dispense with the idea of disorder altogether as it relates to entropy. I think it’s generally more confusing than helpful.
But the technical, quantifiable definition of entropy is a measure of the number of accessible microstates in a system that are macroscopically indistinguishable.
S = k ln W
Entropy S has units of energy divided by temperature, I’ll use units of J/K. The Boltzmann constant k is the constant 1.38 x 10-23 J/K. The Boltzmann constant has the same units as entropy so those will cancel, leaving W as just a number with no dimensions.
W is the number of accessible microstates in a system that are macroscopically indistinguishable. So we need to talk about macrostates and microstates. An example of a macrostate is the temperature and pressure of a system. The macrostate is something we can measure with our instruments: temperature with a thermometer and pressure with a pressure gauge. But at the microscopic or molecular level the system is composed of trillions of molecules and it’s the motion of these molecules that produce what we see as temperature and pressure at a macroscopic level. The thermal energy of the system is distributed between its trillions of molecules and every possible, particular distribution of thermal energy between each of these molecules is an individual microstate. The number of ways that thermal energy of a system can be distributed among its molecules is an unfathomably huge number. But the vast majority of them make absolutely no difference at a macroscopic level. The vast majority of the different possible microstates correspond to the same macrostate and are macroscopically indistinguishable.
To dig a little further into what this looks like at the molecular level, the motion of a molecule can take the form of translation, rotation, and vibration. Actually, in monatomic molecules it only takes the form of translation, which is just its movement from one position to another. Polyatomic molecules can also undergo rotation and vibration, with the number of vibrational patterns increasing as the number of atoms increases and shape of the molecule becomes more complicated. All these possibilities for all the molecules in a system are potential microstates. And there’s a huge number of them. Huge, but also finite. A fundamental postulate of quantum mechanics is that energy is quantized. Energy levels are not continuous but actually come in discrete levels. So there is a finite number of accessible microstates, even if it’s a very huge finite number.
For a system like a piston we can set its entropy by setting its energy (U), volume (V), and number of atoms (N); its U-V-N conditions. If we know these conditions we can predict what the entropy of the system is going to be. The reason for this is that these conditions set the number of accessible microstates. The reason that the number of accessible microstates would correlate with the number of atoms and with energy should be clear enough. Obviously having more atoms in a system will make it possible for that system to be in more states. The molecules these atoms make up can undergo translation, rotation, and vibration and more energy makes more of that motion happen. The effect of volume is a little less obvious but it has to do with the amount of energy separating each energy level. When a set number of molecules expand into a larger volume the energy difference between the energy levels decreases. So there are more energy levels accessible for the same amount of energy. So the number of accessible microstates increases.
The entropies for many different substances have been calculated at various temperatures and pressures. There’s especially an abundance of data for steam, which has had the most practical need for such data in industry. Let’s look at some examples with water at standard pressure and temperature conditions. The entropy of
Solid Water (Ice): 41 J/mol-K
Liquid Water: 69.95 J/mol-K
Gas Water (Steam): 188.84 J/mol-K
One mole of water is 18 grams. So how many microstates does 18 grams of water have in each of these cases?
First, solid water (ice):
S = k ln W
41 J/K = 1.38 x 10-23 J/K * ln W
Divide 41 J/K by 1.38 x 10-23 J/K and the units cancel
ln W = 2.97 x 1024
That’s already a big number but we’re not done yet.
Raise e (about 2.718) to the power of both sides
W = 10^(1.29 x 10^24) microstates
W = 101,290,000,000,000,000,000,000,000 microstates
That is an insanely huge number.
Using the same method, the value for liquid water is:
W = 10^(2.2 x 10^24) microstates
W = 102,200,000,000,000,000,000,000,000 microstates
And the value for steam is:
W = 10^(5.94 x 10^24) microstates
W = 105,940,000,000,000,000,000,000,000 microstates
In each case the increased thermal energy makes additional microstates accessible. The fact that these are all really big numbers makes it a little difficult to see that, since these are differences in exponents, each number is astronomically larger than the previous one. Liquid water has 10^(9.1 x 10^23) times as many accessible microstates as ice. And steam has 10^(3.74 x 10^24) times as many accessible microstates as liquid water.
With these numbers in hand let’s stop a moment to think about the connection between entropy and probability. Let’s say we set the U-V-N conditions for a system of water such that it would be in the gas phase. So we have a container of steam. We saw that 18 grams of steam has 10^(5.94 x 10^24) microstates. The overwhelming majority of these microstates are macroscopically indistinguishable. In most of the microstates the distribution of the velocities of the molecules is Gaussian; they’re not all at identical velocity but they are distributed around a mean along each spatial axis. That being said, there are possible microstates with different distributions. For example, there are 10^(1.29 x 10^24) microstates in which that amount of water would be solid ice. That’s a lot! And they’re still accessible. There’s plenty of energy there to access them. And a single microstate for ice is just as probable as a single microstate for steam. But there are 10^(4.65 x 10^24) times as many microstates for steam than there are for ice. It’s not that any one microstate for steam is more probable than any one microstate for ice. It’s just that there are a lot, lot more microstates for steam. The percentage of microstates that take the form of steam is not 99% or 99.99%. It’s much, much closer than that to 100%. Under the U-V-N conditions that make those steam microstates accessible they will absolutely dominate at equilibrium.
What if we start away from equilibrium? Say we start our container with half ice and half steam by mass. But with the same U-V-N conditions for steam. So it has the same amount of energy. What will happen? The initial conditions won’t last. The ice will melt and boil until the system just flips among the vast number of microstates for steam. If the energy of the system remains constant it will never return to ice. Why? It’s not actually absolutely impossible in principle. But it’s just unimaginably improbable.
That’s what’s going on at the molecular level. Macroscopically entropy is a few levels removed from tangible, measured properties. What we see macroscopically are relations between heat flow, temperature, pressure, and volume. But we can calculate the change in entropy between states using various equations expressed in terms of these macroscopic properties that we can measure with our instruments.
For example, we can calculate the change in entropy of an ideal gas using the following equation:
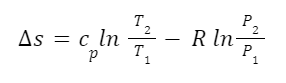
Here s is entropy, cp is heat capacity at constant pressure, T is temperature, R is the ideal gas constant, and P is pressure. We can see from this equation that, all other things being equal, entropy increases with temperature and decreases with pressure. And this matches what we saw earlier. Recall that if the volume of a system of gas increases with a set quantity of material the energy difference between the energy levels decreases and there are more energy levels accessible for the same amount of energy. Under these circumstances pressure would decrease so entropy would decrease with pressure.
For solids and liquids we can assume that they are incompressible and leave off the pressure terms. So the change in entropy for a solid or liquid is given by the equation:
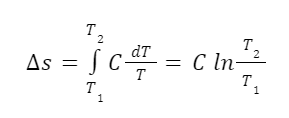
Let’s do an example with liquid water. What’s the change in entropy, and the increase in the number of accessible microstates, that comes from increasing the temperature of liquid water one degree Celsius? Let’s say we’re increasing 1 mole (18 grams) of water from 25 to 26 degrees Celsius. At this temperature the heat capacity of water is 75.3 J/mol-K.

Now that we have the increase in entropy we can find the increase in the number of microstates using the equation

Setting this equal to 0.252 J/mol-K

The increase is not as high as it was with phase changes, but it’s still a very big change.
We’ll wrap up the definition section here but conclude with some general intuitions we can gather from these equations and calculations:
1. All other things being equal, entropy increases with temperature.
2. All other things being equal, entropy decreases with pressure.
3. Entropy increases with phase changes from solid to liquid to gas.
Keeping these intuitions in mind will help as we move to applications with steam
Applications with Steam
The first two examples in this section are thermodynamic cycles. All thermodynamic cycles have 4 processes.
1. Compression
2. Heat addition
3. Expansion
4. Heat rejection
These processes circle back on each other so that the cycle can be repeated. Think, for example, of pistons in a car engine. Each cycle of the piston is going through each of these processes over and over again, several times per second.
There are many kinds of thermodynamic cycles. The idealized cycle is the Carnot cycle, which gives the upper limit on the efficiency of conversion from heat to work. Otto cycles and diesel cycles are the cyles used in gasoline and diesel engines. Our steam examples will be from the Rankine cycle. In a Rankine cycle the 4 processes take the following form:
1. Isentropic compression
2. Isobaric heat addition
3. Isentropic expansion
4. Isobaric heat rejection
An isobaric process is one that occurs at constant pressure. An adiabatic process is one that occurs at constant entropy.
An example of a Rankine cycle is a steam turbine or steam engine. Liquid water passes through a boiler, the steam passes through a turbine, expanding and turning the turbine, The fluid passes through a condenser, and then is pumped back to the boiler, where the cycle repeats. In such problems the fact that entropy is the same before and after expansion through the turbine reduces the number of unknown variables in our equations.
Let’s look at an example problem. Superheated steam at 6 MPa at 600 degrees Celsius expands through a turbine at a rate of 2 kg/s and drops in pressure to 10 kPa. What’s the power output from the turbine?
We can take advantage of the fact that the entropy of the fluid is the same before and after expansion. We just have to look up the entropy of superheated steam in a steam table. The entropy of steam at 6 MPa at 600 degrees Celsius is:
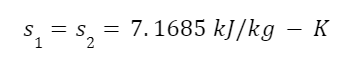
The entropy of the fluid before and after expansion is the same but some of it condenses. This isn’t good for the turbines but it happens nonetheless. Ideally, most of the fluid is still vapor so the ratio of the mass that is saturated vapor to the total fluid mass is called “quality”. The entropies of saturated liquid, sf, and of evaporation, sfg, are very different. So we can use algebra to calculate the quality, x2, of the fluid. The total entropy of the expanded fluid is given by the equation:

s2 we already know because the entropy of the fluid exiting the turbine is the same as that of the fluid entering the turbine. And we can look up the other values in steam tables.

Solving for quality we find that
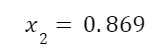
Now that we know the quality we can find the work output from the turbine. The equation for the work output of the turbine is:
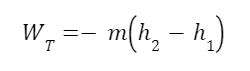
h1 and h2 and enthalpies before and after expansion. If you’re not familiar with enthalpy don’t worry about it (we’re getting into enough for now). It roughly corresponds to the substance’s energy. We can look up the enthalpy of the superheated steam in a steam table.

For the fluid leaving the turbine we need to calculate the enthalpy using the quality, since it’s part liquid, part vapor. We need the enthalpy of saturated liquid, hf, and of evaporation, hfg. The total enthalpy of the fluid leaving the turbine is given by the formula

From the steam tables
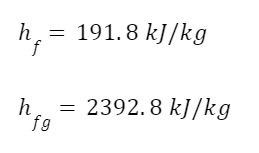
So
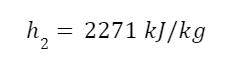
And now we can plug this in to get the work output of the turbine.

So here’s an example where we used the value of entropy to calculate other observable quantities in a physical system. Since the entropy was the same before and after expansion we could use that fact to calculate the quality of the fluid leaving the turbine, use quality to calculate the enthalpy of the fluid, and use the enthalpy to calculate the work output of the turbine.
A second example. Superheated steam at 2 MPa and 400 degrees Celsius expands through a turbine to 10 kPa. What’s the maximum possible efficiency from the cycle? Efficiency is work output divided by heat input. We have to input work as well to compress the fluid with the pump so that will subtract from the work output from the turbine. Let’s calculate the work used by the pump first. Pump work is:
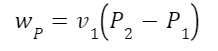
Where v is the specific volume of water, 0.001 m3/kg. Plugging in our pressures in kPa:

So there’s our pump work input.
The enthalpy of saturated liquid is:
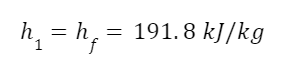
Plus the pump work input is:

Now we need heat input. The enthalpy of superheated steam at 2 MPa and 400 degrees Celsius is:

So the heat input required is:

The entropy before and after expansion through the turbine is the entropy of superheated steam at 2 MPa and 400 degrees Celsius is:

As in the last example, we can use this to calculate the quality of the steam with the equation:
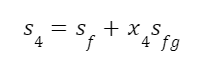
Looking up these values in a steam table:

Plugging these in we get:
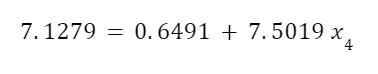
And

Now we can calculate the enthalpy of the expanded fluid.
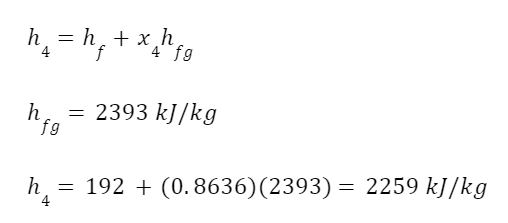
And the work output of the turbine.
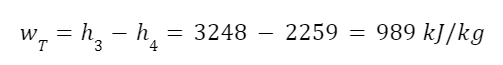
So we have the work input of the pump, the heat input of the boiler, and the work output of the turbine. The maximum possible efficiency is:
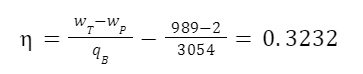
So efficiency is 32.32%.
Again, we used entropy to get quality, quality to get enthalpy, enthalpy to get work, and work to get efficiency. In this example we didn’t even need the mass flux of the system. Everything was on a per kilogram basis. But that was sufficient to calculate efficiency.
One last example with steam. The second law of thermodynamics has various forms. One form is that the entropy of the universe can never decrease. It is certainly not the case that entropy can never decrease at all. Entropy decreases all the time within certain systems. In fact, all the remaining examples in this episode will be cases in which entropy decreases within certain systems. But the total entropy of the universe cannot decrease. Any decrease in entropy must have a corresponding increase in entropy somewhere else. It’s easier to see this in terms of an entropy balance.

The entropy change in a system can be negative but the balance of the change in system entropy, entropy in, entropy out, and entropy of the surroundings will never be negative. We can look at the change of entropy of the universe as a function of the entropy change of a system and the entropy change of the system’s surroundings.
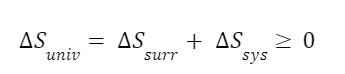
So let’s look at an example. Take 2 kg of superheated steam at 400 degrees Celsius and 600 kPa and condense it by pulling heat out of the system. The surroundings have a constant temperature of 25 degrees Celsius. From steam tables the entropy of the superheated steam and saturated steam are:
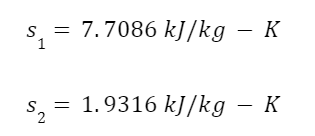
With these values we can calculate the change in entropy inside the system using the following equation;

The entropy decreases inside the system. Nothing wrong with this. Entropy can definitely decrease locally. But what happens in the surroundings? We condensed the steam by pulling heat out of the system and into the surroundings. So there is positive heat flow, Q, out into the surroundings. We can find the change in entropy in the surroundings using the equation:
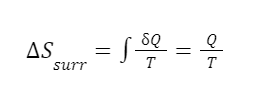
We know the surroundings have a constant temperature, so we know T. We just need the heat flow Q. We can calculate the heat flow into the surroundings by calculating the heat flow out of the system using the equation

So we need the enthalpies of the superheated steam and saturated steam.

And plugging these in
Q = mΔh=(2)3270.2-670.6=5199 J
Now that we have Q we can find the change in entropy in the surroundings:

The entropy of the surroundings increases. And the total entropy change of the universe is:
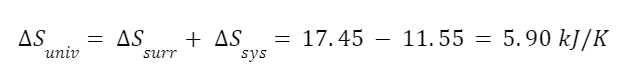
So even though entropy decreases in the system the total entropy change in the universe is positive.
I like these examples with steam because they’re very readily calculable. The thermodynamics of steam engines have been extensively studied for over 200 years, with scientists and engineers gathering empirical data. So we have abundant data on entropy values for steam in steam tables. I actually think just flipping through steam tables and looking at the patterns is a good way to get a grasp on the way entropy works. Maybe it’s not something you’d do for light reading on the beach but if you’re ever unable to fall asleep you might give it a try.
With these examples we’ve looked at entropy for a single substance, water, at different temperatures, pressures, and phases, and observed the differences of the value of entropy at these different states.
To review some general observations:
1. All other things being equal, entropy increases with temperature.
2. All other things being equal, entropy decreases with pressure.
3. Entropy increases with phase changes from solid to liquid to gas.
In the next section we’ll look at entropies for changing substances in chemical reactions.
Applications with Chemical Reactions
The most important equation for the thermodynamics of chemical reactions is the Gibbs Free Energy equation:
ΔG=ΔH-TΔS
Where H, T, S are enthalpy, temperature, and entropy. ΔG is the change in Gibbs free energy. Gibbs free energy is a thermodynamic potential. It is minimized when a system reaches chemical equilibrium. For a reaction to be spontaneous the value for ΔG has to be negative, meaning that during the reaction the Gibbs free energy is decreasing and moving closer to equilibrium.
We can see from the Gibbs free energy equation
ΔG=ΔH-TΔS
That the value of the change in Gibbs free energy is influenced by both enthalpy and entropy. The change in enthalpy tells us whether a reaction is exothermic (negative ΔH) or endothermic (positive ΔH). Exothermic reactions release heat while endothermic reactions absorb heat. This has to do with the total change in the chemical bond energies in all the reactants against all the products. In exothermic reactions the energy released from breaking chemical bonds is greater than the energy used to form new chemical bonds. This extra energy is converted to heat. We can see from the Gibbs free energy equation that exothermic reactions are more thermodynamically favored. Nevertheless, entropy can override enthalpy.
The minus sign in front of the TS term tells us that an increase in entropy where ΔS is positive will be more thermodynamically favored. This makes sense with what we know about entropy from the second law of thermodynamics and from statistical mechanics. The effect is proportional to temperature. At low temperatures entropy won’t have much influence and enthalpy will dominate. But at higher temperatures entropy will start to dominate and override enthalpic effects. This makes it possible for endothermic reactions to proceed spontaneously. If the increase in entropy for a chemical reaction is large enough and the temperature is high enough endothermic reactions can proceed spontaneously, even though the energy required to form the chemical bonds of the products is more than the energy released from the chemical bonds in the reactants.
Let’s look at an example. The chemical reaction for the production of water from oxygen and hydrogen is:
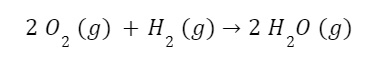
We can look up the enthalpies and entropies of the reactants and products in chemical reference literature. What we need are the standard enthalpies of formation and the standard molar entropies of each of the components.
The standard enthalpies of formation of oxygen and hydrogen are both 0 kJ/mol. By definition, all elements in their standard states have a standard enthalpy of formation of zero. The standard enthalpy of formation for water is -241.83 kJ/mol. The total change in enthalpy for this reaction is
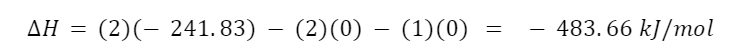
It’s negative which means that the reaction is exothermic and enthalpically favored.
The standard molar entropies for hydrogen, oxygen, and water are, respectively, 130.59 J/mol-K, 205.03 J/mol-K, and 188.84 J/mol-K. The total change in entropy for this reaction is

It’s negative so entropy decreases in this reaction, which means the reaction is entropically disfavored. So enthalpy and entropy oppose each other in this reaction. Which will dominate depends on temperature? At 25 degrees Celsius (298 K) the change in Gibbs free energy is

The reaction is thermodynamically favored. Even though entropy is reduced in this reaction, at this temperature that effect is overwhelmed by the favorable reduction in enthalpy as chemical bond energy of the reactants is released as thermal energy.
Where’s the tradeoff point where entropy overtakes enthalpy? This is a question commonly addressed in polymer chemistry with what’s called the ceiling temperature. Polymers are macromolecules in which smaller molecular constituents called monomers are consolidated into larger molecules. We can see intuitively that this kind of molecular consolidation constitutes a reduction in entropy. It corresponds with the rough analogy of greater order from “disorder” as disparate parts are assembled into a more organized totality. And that analogy isn’t bad. So in polymer production it’s important to run polymerization reactions at temperatures where exothermic, enthalpy effects dominate. The upper end of this temperature range is the ceiling temperature.
The ceiling temperature is easily calculable from the Gibbs free energy equation for polymerization

Set ΔGp to zero.

And solve for Tc

At this temperature enthalpic and entropic effects are balanced. Below this temperature polymerization can proceed spontaneously. Above this temperature depolymerization can proceed spontaneously.
Here’s an example using polyethylene. The enthalpies and entropies of polymerization for polyethylene are
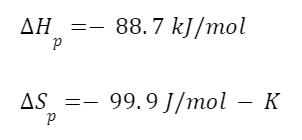
Using our equation for the ceiling temperature we find

So for a polyethylene polymerization reaction you want to run the reaction below 610 degrees Celsius so that the exothermic, enthalpic benefit overcomes your decrease in entropy.
Conclusion
A friend and I used to get together on weekends to take turns playing the piano, sight reading music. We were both pretty good at it and could play songs reasonably well on a first pass, even though we’d never played or seen the music before. One time when someone was watching us she asked, “How do you do that?” My friend had a good explanation I think. He explained it as familiarity with the patterns of music and the piano. When you spend years playing songs and practicing scales you just come to know how things work. Another friend of mine said something similar about watching chess games. He could easily memorize entire games of chess because he knew the kinds of moves that players would tend to make. John Von Neumann once said: “In mathematics you don’t understand things. You just get used to them.” I would change that slightly to say that you understand things by getting used to them. Also true for thermodynamics. Entropy is a complex property and one that’s not easy to understand. But I think it’s easiest to get a grasp on it by using it.

